MFDFF Menu
Molecular Fragments Described Force Field
Database
This command opens a dialog that shows the content of MFDFF database, and provides tools for managing the database.
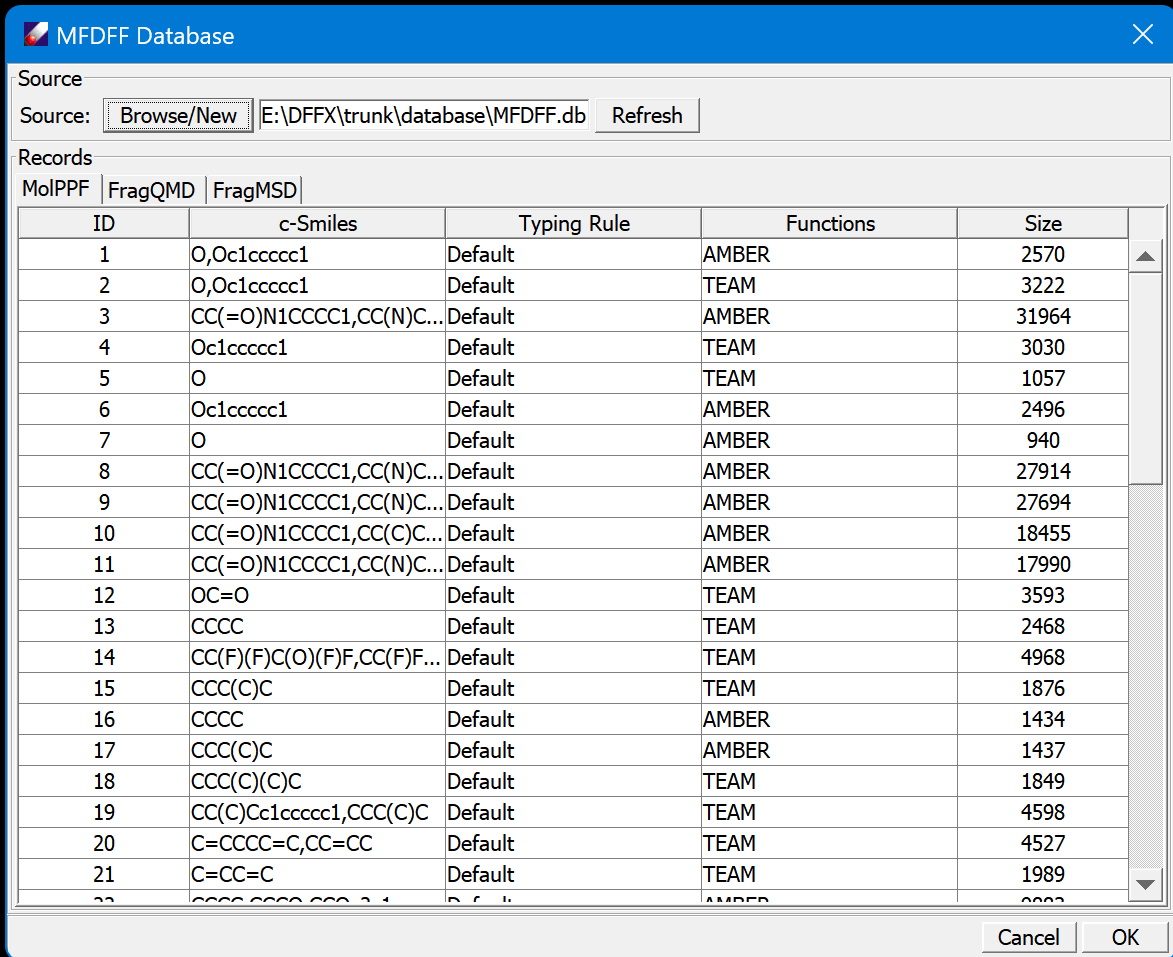
The top section "Source" is for managing the database file. The button Browse/New is a command to create or open a database, which is a binary file can be placed in any places. The default location is in <dff-root>\database. Refresh updates the contents of the tables.
The database has three tables, MolPPF, FragQMD, and FragMSD. MolPPF is a table of molecule type force fields, FragQMD is for QMD files of fragments, and FragMSD contains MSD files of fragments to be calculated. The contents of FragMSD are changed dynamically, the fragments will be removed if their QMD files are updated.
Mouse left-click on the tab to change the displaying table. Mouse left-click on the head of column to sort the data.
In each table, select one or multiple rows and mouse right-click will open a popup command, which contains Download, Delete for all three tables, and Compute for FragMSD. Download is used to download the selected records. Delete is used for removing the selected entries. Note this operation is not reversible, make sure the database is backed up before deleting any entries. Compute is used to compute the selected fragments.
The following commands are designed for assign force fields for molecular models in steps.
Enquiry Force Field
This command applies to any molecule models, large or small. One or multiple models can be handled at once.
Select the model(s) from Project Navigator, click on this command to open a dialog. The dialog lists the selected files. Click Cancel to quit. OK to start the computation. A new Computation window appears to show the output of the background job.
In the background, DFF analyzes each of the models, identifies molecule types in the models, and searches the MolFF table for force fields of the molecule types. If all required force fields are obtained, an integrated force field will be made for the model. If any molecule type force fields are missing, the corresponding molecule types and their molecular fragments will be saved in subdirectories named as "MolType" and "Fragment" respectively for parameterization.
Collect Fragment QMD
Select the fragments to be parametrized and open this command to start a background job to search and download corresponding QMD files from the FragQMD table. Depending on the results of searching, several options can be takenL: for fragments without QMD saved, either compute the data or save the fragments for later (batch) computations; for fragments already have QMD in the database, either recompute the data or skip new computation. The fragments to be calculated will be saved in FragMSD table.

This dialog is used to set up QMD computation options. The jobs can be run in DFF on the local host computer, or saved for later. The computation is done by using Gaussian program package, driven by DFFQMD.exe program. The Task section provides selection of what to be computed.
Build MolType PPF
When all required QMD files are in the database, use this command to build force fields for molecule types. For each molecule type, DFF searches and downloads required fragment QMD files, then starts the regression using a default fitting and validation procedure. When the job is done, a summary of regressions will be displayed. Details can be found in output (.dfo) files.
Assign and Test MolType PPF
This command is used to assigns the molecule type force fields obtained from the regression of the fragment data of the molecule type. This assignment will confirm the force field is complete for the molecule, and remove any redundant parameters due to the overlaps among the fragments. In addition, DFF will run a short MD simulation to test the stability of the result force field. If all appears to be right, prompt to update the MolPPF table. When all required molecule type force fields are developed and uploaded, repeat Enquiry command to assign force fields for the target models.
The above steps are integrated into one workflow command.
WorkFlow
This is an automated workflow for assigning force fields for target molecular models automatically. For a set of models, the workflow searches or developes required molecular type force fields and build integrated force fields for the models. The new force field type force fields are developed when they are in need, the workflow builds up the collection of molecule type force fields in a self-leaning manner.
This command opens a dialog to set parameters for the workflow:
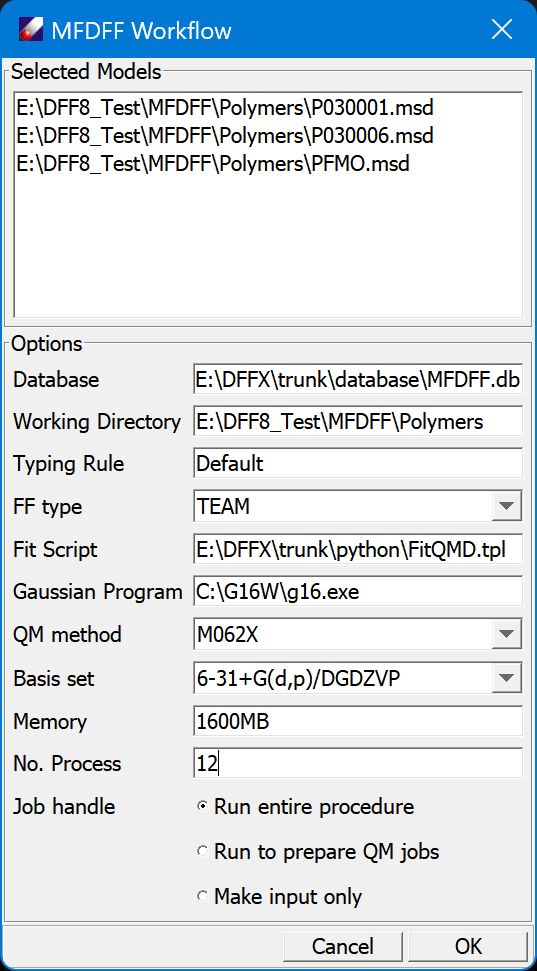
The selected models are listed on the top section.
"Database" is the database selected. Use MFDFF/Database command to change the database. The typing rule is fixed by the database because it determines the partitions of fragments. You may choose "FF type". The "Fit Script" is a text file used to generate fitting input. This file can be edited so that you may optimize the fitting procedure. You must set the "Gaussian program" properly. "QM Method" and "Basis set" can be changed, note the default is used for building the default database. "Memory" and "No. Process" need to be set according to your computer setup. These options imply to use the Multi-Thread Parallel (MTP) method, which means using multiple threads on one computer. The last section set how the program to run. "Run entire procedure" will do all computations inside of DFF, which may take a long time. "Collect fragments for QM Jobs" will prepare QM jobs for the fragments to be calculated and save the fragments in the FragMSD table; you may do the QM jobs and update the database later. "Make an input file only" will make an input file for the workflow, you can execute it without DFF interface.
In addition, the following commands are a collection of tools, which can be used to accomplish specific tasks.
Molecule Type
This command works on any models, it is used to find the molecular types in the models and save the result in /MolType folder.
Standard Fragment
This works on molecular types, identify constituent fragments and save the fragments in /Fragment folder.
Update FragQMD
This command uploads the selected QMD files to FragQMD table. If any QMD files exist in the table, identified by fragment SMILES strings, the existing ones will be overwritten. If update is done successfully, corresponding fragments saved in FragMSD table will be removed.
Download FragQMD
This command uses fragment MSD to search and download corresponding QMD.
Update MolPPF
Load PPF files for molecule types, identified as molecule type MSD files.
Download MolPPF
Search and download PPF files for molecule types, provided as MSD files.
Program Fit
This command provides flexible fitting options, organized as multiple tasks, each is presented by a tabbed panel. The jobs can be run sequentially or independently, so that different options can be tested and compared.